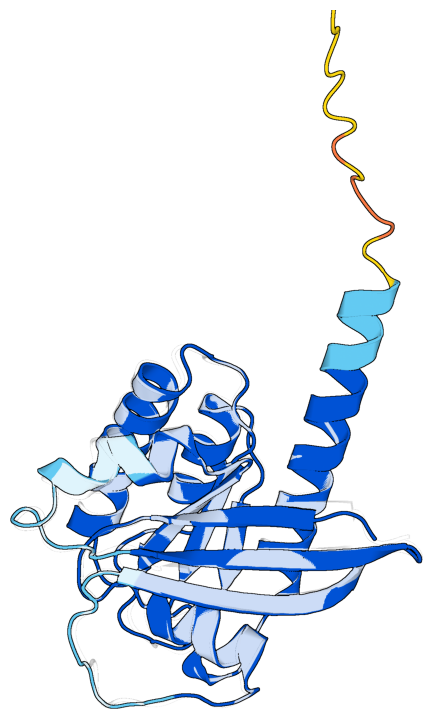
Compare structures with pLDDT coloring#
Superimpose an AlphaFold prediction onto an experimental structure and render both together, with the prediction colored by per-residue pLDDT.
portein.superimpose_by_alignment()— pairs CA atoms between two structures by re-aligning each structure’s CA-derived sequence to the input alignment, so structures with missing residues are handled transparently.portein.by_plddt()— bins residues into AlphaFold’s standard confidence bands, returning a dict shaped forProteinConfig.highlight_residues.
import tempfile
from pathlib import Path
import matplotlib.pyplot as plt
import yaml
from biotite.sequence import ProteinSequence
from biotite.sequence.align import SubstitutionMatrix, align_optimal
from PIL import Image
import portein
output_dir = Path(tempfile.mkdtemp())
Load the structures#
7lc2 is a crystal structure of KRAS bound to a non-hydrolyzable GTP
analog (GNP). AF_P01116 is the AlphaFold prediction for the same
UniProt entry.
crystal = portein.read_structure("../_data/7lc2.pdb")
af_model = portein.read_structure("../_data/AF_P01116.cif")
crystal_chain_a = crystal[crystal.chain_id == "A"]
Align the input sequences#
Build a pairwise alignment of the crystal chain A sequence against the
AlphaFold prediction. superimpose_by_alignment will then re-align
the structure-derived sequences to this input alignment internally,
so missing residues in the crystal structure cause no surprises.
def to_sequence(structure):
ca = structure[structure.atom_name == "CA"]
return ProteinSequence("".join(ProteinSequence.convert_letter_3to1(r) for r in ca.res_name))
crystal_seq = to_sequence(crystal_chain_a)
af_seq = to_sequence(af_model)
alignment = align_optimal(crystal_seq, af_seq, SubstitutionMatrix.std_protein_matrix())[0]
Superimpose and bin by pLDDT#
superimpose_by_alignment returns the rotated AlphaFold model
combined with the crystal structure into one AtomArray. by_plddt
reads the AlphaFold b_factor column (where AlphaFold stores pLDDT)
and bins each residue into AlphaFold’s standard color bands.
combined, _, paired = portein.superimpose_by_alignment(
query=crystal_chain_a,
target=af_model,
alignment=alignment,
query_chain="A",
target_chain_id="B",
query_chain_id="A",
)
# Color the *target* (now chain "B" in `combined`) by pLDDT.
af_in_combined = combined[combined.chain_id == "B"]
highlight = portein.by_plddt(af_in_combined)
print(f"Superposed using {paired.shape[0]} CA pairs.")
Superposed using 166 CA pairs.
Render the composite#
The crystal is plain white but partially transparent so the pLDDT-
colored AlphaFold prediction underneath stands out. Per-chain
chain_transparency uses PyMOL’s depth-aware cartoon_transparency
setting, so the two structures composite correctly per-pixel rather
than as flat 2D layers.
with open("../../configs/pymol_settings.yaml") as f:
pymol_settings = yaml.safe_load(f)
protein_config = portein.ProteinConfig(
pdb_file=combined,
rotate=False,
width=600,
chain_to_color={"A": "white", "B": "lightgray"},
highlight_residues=highlight,
chain_transparency={"A": 0.2},
output_prefix=str(output_dir / "compare"),
)
image_file = portein.Pymol(
protein=protein_config,
layers=[
portein.PymolConfig(
representation="cartoon",
pymol_settings=pymol_settings,
selection="all",
),
],
buffer=2,
).run()
Crop and display#
cropped = portein.crop_to_content(Image.open(image_file))
DPI = 100
fig, ax = plt.subplots(figsize=(cropped.width / DPI, cropped.height / DPI), dpi=DPI)
ax.imshow(cropped)
ax.axis("off")
fig.subplots_adjust(left=0, right=1, top=1, bottom=0)
plt.show()